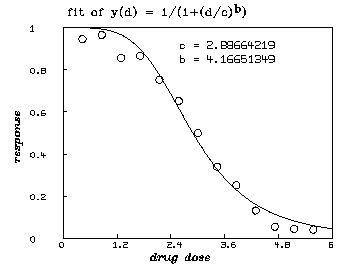
Suppose we have a set of drugs (or other treatments which we misclassify as ``drugs'' for the purpose of our analysis.) These drugs are supposed to be treatments for some disease, e.g. , HIV infection. Our goal is to assess whether, and to what extent, treatment with a particular combination of these drugs is more or less effective than some other such combination treatment. Such combination treatments include the case of treatment with a single drug, so we can also compare the efficacy of two distinct single drugs with our method.
For each treatment with a combination of drugs , the data we have is a set of dimensional points of the form , where is the dose of drug (measured in any desirable units) and is the response observed in the subject represented by the point at hand.
The observed response is always non-negative, and is generally a value in the interval . Such a value denotes the relative amount or level of disease present after the drug treatment. Thus a small response value indicates a better response than does a larger response value. Crudely, a response value should be measured such that is the percent improvement observed. For example, let denote a subject's white blood cell count before treatment and let denote that subject's white blood cell count after treatment; then the response for that subject might be which is 1 minus the precentage improvement observed. The value 1 or greater denotes no suppression of disease, and values near 0 indicate substantial suppression of disease. Another example is that where we measure a quantity that is directly related to the level of disease present, such as the concentration of PSA present, normalized to lie in the interval , although here, as in most cases, a percentage response is more appropriate, since the amount of disease present prior to treatment can vary greatly.
For a single drug, a so-called logistic dose-response curve often provides an adequate description of the effect of treatment with that drug. We assume that such a model for single drugs is appropriate here. (Although, see the remarks below on a non-parametric approach.) The general logistic dose-response function for a single drug is:
In our case, we assume that ( i.e. , we have no suppression of disease at 0 dose) and that ( i.e. , we approach complete suppression of disease as the dose becomes sufficiently large.) Thus, . This dose-response curve generally ``decays'' sigmoidally from the value 1 to the value 0 as the dose increases. If , we have faster decay, if , we have slower decay, if , is the constant value .5, and if , our dose-response curve inceases instead of decaying.
We may construct a single drug dose-response model for given
dose-response data by estimating
the parameters and that
fit the model function to
our data via weighted least-squares minimization. An example showing the
use of the MLAB mathematical and statistical modeling system[1]
to read-in the dose-response data,
define the single-drug logistic dose-response model function,
provide initial guesses for
the parameters and , fit
our model to the data to obtain the
least-squares estimates of and ,
and finally, to graph the results,
is given below. (Often it is necessary to impose constraints on the
parameters and in
order to get a successful fit.
This can be done in MLAB, but it is not
needed in the example given here.)
* dv=read(d1data,100,2)
* fct y(d) = 1/(1+(d/c)^b)
* c = 3; b = 1
* fit (b,c), y to dv
final parameter values
value error dependency parameter
4.166513485 0.3632194177 0.05378619542 B
2.886642188 0.06692784189 0.05378619542 C
6 iterations
CONVERGED
best weighted sum of squares = 2.664797e-02
weighted root mean square error = 4.921934e-02
weighted deviation fraction = 5.824418e-02
R squared = 9.833524e-01
* draw points(y,0:6!100)
* draw dv lt none pt circle ptsize .02
* left title "response" font 18
* bottom title "drug dose" font 18
* top title "fit of y(d) = 1/(1+(d/c)'.5ub'.5d)" font 7
* title "c = "+c at (.5,.8)
* title "b = "+b at (.5,.75)
* view
Now let us consider a two-drug combination treatment. The dose-response model in this situation is a function of two arguments, , that predicts the response to the combination dose . Let us suppose that drug and drug have no interaction. Moreover, we postulate that and . (The '^' symbol denotes the exponentiation operation; we must use it due to the inadequacies of HTML.) We assume that the parameters , , , and are known due to fitting the single-drug logistic dose-response model to given single-drug dose-response data for drug and separately for drug . In analogy to the terminology used with multivariate distribution functions, we may call the single-drug logistic functions and the marginal dose-response functions for the two-argument dose-response function .
Let be the value such that our non-interaction two-drug dose-response model satisfies , and let be the value such that . Now based on our no-interaction hypothesis, we can follow Bunow and Weinstein [2], and geometrically define by postulating that for on the line-segment connecting and . Note it would not be appropriate to define since drug and drug compete to suppress the disease, even if they act independently; that is, when drug has acted to suppress some of the disease, there is less ``disease'' present for drug to apply to, and conversely. The line-segment connecting and is , and we specify that for belongs to the set . This is appropriate because, when drug is the same as drug , the predicted response to a combination dose must be the same as the predicted response to a dose of size of drug (or ) alone! Any drug-interaction model which fails to satisfy this condition cannot be a statistically-correct model.
Now note that, if , then .
Recall that . Thus , so . Also , so , and thus .
But then, when belongs to the set , and for some value such that , and we have specified that . Next, note that
This Bunow-Weinstein two-argument dose-response function for
non-interacting drugs is
a logically-correct generalization of a single-drug
logistic dose-response function. This implicit function
can be defined in MLAB as shown below. Note the care that is
taken to avoid division by zero and raising zero to a negative power.
function y1(d1)=1/(1+(d1/c1)^b1)
function y2(d2)=1/(1+(d2/c2)^b2)
function y(d1,d2)=if d1=0 then y2(d2) else
if d2=0 then y1(d1) else
if y1(d1)<.00001 and y2(d2)<.00001 then 0 else
if y1(d1)>.99990 and y2(d2)>.99990 then 1 else
root(z,1e-11,1-1e-11,(d1/c1)*(1/z-1)^(-1/b1)+(d2/c2)*(1/z-1)^(-1/b2) -1)
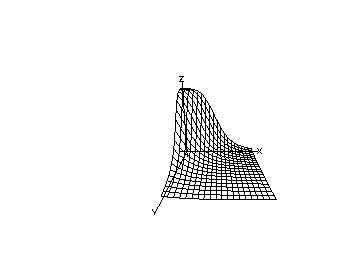
The MLAB commands used to produce a
picture of the ``response-surface'' defined by our model
function for two
non-interacting drugs with identical marginal curves
defined by
and are given below.
We assume the commands defining the functions
, ,
and
have been executed.
/* define c1=c2=2.8866 and b1=b2=4.1665 */
c1=2.8866; c2=c1; b1=4.1665; b2=b1
/* draw a 3D perspective view of the non-interaction drug response
surface for two drugs having the same marginal single drug
dose-response curves. */
s = cross(0:6!20,0:6!20)
s col 3 = y on s
draw s linetype net
cmd3d("axes")
view
The produced picture is shown below.
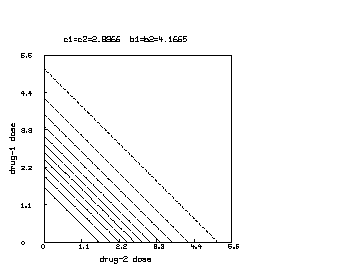
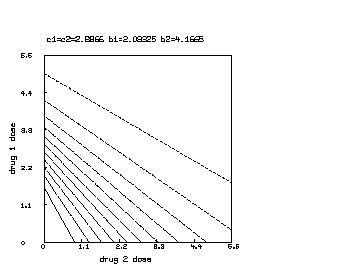
We may continue the above MLAB dialog to produce a contour map corresponding to the function with and corresponding to the surface shown above. The contour map shows some level lines where is constant.
delete w3 /* draw a contour map corresponding to the surface above. */ draw contour(s) linetype vmarker left title "drug-1 dose" bottom title "drug-2 dose" top title "c1=c2="+c+" b1=b2="+b view
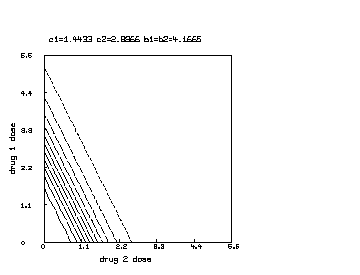
We may further continue the above MLAB dialog to produce a contour map corresponding to the function in the case where , and .
delete w /* draw a contour map of the non-interaction combination drug response surface where c1 is reduced to half of c2. */ c1 = c1*.5 s = cross(0:6!20,0:6!20) s col 3 = y on s draw contour(s) linetype vmarker left title "drug 1 dose" bottom title "drug 2 dose" top title "c1="+c1+" c2="+c2+" b1=b2="+b1 view
Finally, we may further continue the above MLAB dialog to produce a contour map corresponding to the function in the case where , and .
delete w /* draw a contour map of the non-interaction combination drug response surface where b1 is reduced to half of b2. */ c1 = c2; b1 = b1*.5 s col 3 = y on s col 1:2 draw contour(s) linetype vmarker left title "drug 1 dose" bottom title "drug 2 dose" top title "c1=c2="+c+" b1="+b1+" b2="+b2 view
Note that the Bunow-Weinstein non-interacting two-drug dose-response function can be immediately generalized to the case of non-interacting drugs. In general we have:
Now we may consider drug combination treatments where the drugs involved may interact synergistically or antagonistically. To analyze such treatments, we need to have estimated the parameters , , , , appearing in the marginal single-drug dose-response functions for the drugs under consideration, so that the Bunow-Weinstein non-interaction model is specified.
Given the data for , we may compute the predicted non-interaction response values . If we have no interaction, each value should be approximately the same as the corresponding observed response value . If not, then we can conclude there is evidence for a synergistic or antagonistic effect, and the size and direction of this effect can be estimated from the response differences . (Note that for the same combination of drugs, we might have both synergistic effects for dose-pairs in some regions and antagonistic effects in different dose-pair regions.)
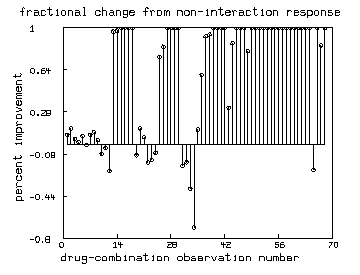
It may be convenient to consider the fractional differences ; is the percent change from the non-interaction predicted response that is observed in the actual response . If , the disease was suppressed more than the non-interaction model would predict and . If , the disease was suppressed less than the non-interaction model would predict and . Crudely, we might say that our drug-combination exhibits a percent mean synergy interaction effect.
It is interesting to plot the -values, the -values and/or the -values versus the associated equivalent single-drug-dose of drug (or any other of the drugs involved) as determined by the Bunow-Weinstein non-interaction model . If is the vector of dose values corresponding to the data-value and the predicted non-interaction value , then the equivalent drug dose is the value such that . An MLAB function involving the root-operator can be defined to compute such equivalent dose values, but can be directly computed as (with due care for and for .) Such plots may be useful in seeing how the synergy effect varies with dose.
Now one way to decide if the -values are, over all, sufficiently greater than the values to conclude that we have statistically-significant synergy is to test the hypothesis that the mean of the -values is less than or equal to 0 versus the alternate hypothesis that the mean of the -values is greater than 0. A similar test can be used to assess whether we have statistically-significant antagonism.
It is probably most appropriate to use a non-parametric test such as the Wilcoxon 2-sample paired-data test in MLAB on the -values versus the -values. And we can check our outcome with a paired-data -test on the -values, even though the normal-distribution assumption used there may be invalid.
Note that in order to decide which of two drug combination treatments is better we need only compare the -values for treatment 1 with the -values for treatment 2.
Let us look at an example using MLAB to perform an analysis of some two drug combination responses at various doses. We have a file called ``a3azt.in'' which contains lines of three numbers, where the first number is the administered dose of drug , the second number is the administered dose of drug ,and the third number is the response measured as the amount of disease present after treatment (in this case, the response is the amount of virus assayed.) Each line in our data file is thus an ``observation'' from a single ``experiment''.
The MLAB dialog given below shows
how we read our data, normalize the response values
to lie in
, extract the data for the marginal dose-response curves
(one data-set where the drug dose is 0 and
one data-set where the drug dose is 0),
fit to obtain the estimated
marginal dose-response curve parameters
, ,
, and
(which then define
our Bunow-Weinstein non-interaction two-drug dose-response
function), compute the predicted non-interaction response
values ,
and then compare the predicted -values
with the observed -values
to study the nature and amount of
synergism apparent between our two drugs.
/* read-in a3azt.in, normalize it, and extract the data needed to
fit the marginals. */
m = read("a3azt.in",1000,3)
m col 3 = (m col 3)/maxv(m col 3)
m1 = extract(m,2,0) col (1,3); m1 = sort(m1)
m2 = extract(m,1,0) col (2,3); m2 = sort(m2)
/* define the non-interacting 2-drug combination model and its marginals */
fct y1(d1) = 1/(1+(d1/c1)^b1)
fct y2(d2) = 1/(1+(d2/c2)^b2)
fct y(d1,d2) = if d1=0 then y2(d2) else
if d2=0 then y1(d1) else
if y1(d1)<.00001 and y2(d2)<.00001 then 0 else
if y1(d1)>.9999 and y2(d2)>.9999 then 1 else
root(z,1e-11,1-1e-11,(d1/c1)*(1/z-1)^(-1/b1)+(d2/c2)*(1/z-1)^(-1/b2)-1)
/* fit the marginals. the derivative y1'b1 involves the log
of d1/c1 which may be 0. Also the derivative y1'c1 involves
(d1/c1)^(b1-1) which may become 0^0. y2 has similar problems.
To avoid these problems we can use symdsw = 0. */
symdsw = 0
constraints q = {b1 > .00001,b2 > .00001,c1 > .00001,c2 > .00001}
c1 = 1; b1 = 1
fit (c1,b1), y1 to m1 with weight ewt(m1), constraints q
final parameter values
value error dependency parameter
0.039470779 0.0063108802 0.05395208237 C1
1.078644618 0.2041472332 0.05395208237 B1
9 iterations
CONVERGED
best weighted sum of squares = 1.028776e+01
weighted root mean square error = 8.572280e-01
weighted deviation fraction = 7.954586e-02
R squared = 9.770071e-01
no active constraints
c2 = 1; b2 = 2
fit (c2,b2), y2 to m2 with weight ewt(m2), constraints q
final parameter values
value error dependency parameter
0.204651635 0.0231668295 0.07182693961 C2
2.676935005 0.6159096057 0.07182693961 B2
11 iterations
CONVERGED
best weighted sum of squares = 4.343704e+01
weighted root mean square error = 1.647669e+00
weighted deviation fraction = 7.585580e-02
R squared = 9.646190e-01
no active constraints
/* graph the resulting y1-fit. */
draw points(y1,minv(m1 col 1):maxv(m1 col 1)!100)
draw m1 lt none pt circle ptsize .01
left title "response" font 18
bottom title "drug-1 dose" font 18
top title "fit of y1(d1) = 1/(1+(d1/c1)'.5ub1'.5d)" font 7
title "c1 = "+c1 at (.5,.8)
title "b1 = "+b1 at (.5,.75)
view
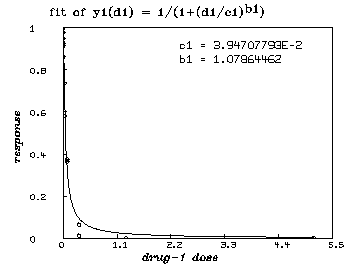
delete w /* graph the resulting y2-fit. */ draw points(y2,minv(m2 col 1):maxv(m2 col 1)!100) draw m2 lt none pt circle ptsize .01 left title "response" font 18 bottom title "drug-2 dose" font 18 top title "fit of y2(d2) = 1/(1+(d2/c2)'.5ub2'.5d)" font 7 title "c2 = "+c2 at (.5,.8) title "b2 = "+b2 at (.5,.75) view
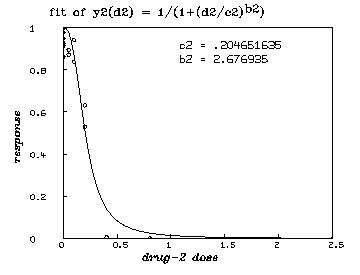
delete w
/* Now to analyze our drug combination data, we compute our non-interaction
response values r, and compare them to the actual data v. If the v's
are smaller, we have synergism. Also we will look at the percent
changed-response values p.*/
r = y on (m col 1:2)
v = m col 3
p = 1-v/'r
/* Type out the average percentage-improvement seen in our
data compared to the predicted non-interaction response. */
type mean(p)
= .593531217
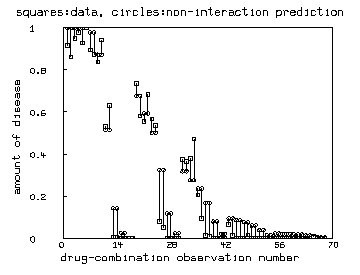
/* Graph the linked-pairs of predicted noninteraction response and
observed response for each drug-dose pair used. */
draw v pt square lt none ptsize .01
draw r pt circle lt none ptsize .01
top title "squares:data, circles:non-interaction prediction"
left title "amount of disease"
bottom title "drug-combination observation number"
n = nrows(v)
nm = 1:n
fct f(x) = if x < 0 then -1 else 1
c = f on (v-r)
qv = nm&'v&'c; qr = nm&'r&'c
qq = mesh(qv,qr)
qv = extract(qq,3,-1) col 1:2
qr = extract(qq,3,1) col 1:2
draw qv lt alternate color green
draw qr lt alternate color red
view
delete w
/* Graph the percentage-improvements (positive or negative) seen
in each observation. */
draw mesh(nm&'0, nm&'p) lt alternate color yellow
draw p pt circle ptsize .01 lt none
draw nm&'0 color red
top title "fractional change from non-interaction response"
bottom title "drug-combination observation number"
left title "percent improvement"
view
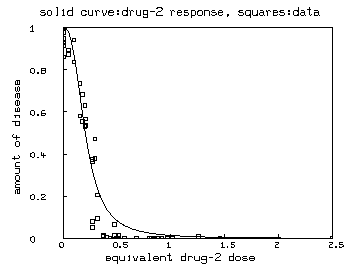
delete w fct ed2(r) = if r=1 then 0 else if r=0 then maxd2 else c2/[(1/r-1)^(-1/b2)] maxd2 = maxv(m col 2) e = ed2 on r draw e&'v color green lt none pt square ptsize .01 /* also show the drug-2 marginal curve */ draw points(y2,minv(m2 col 1):maxv(m2 col 1)!100) top title "solid curve:drug-2 response, squares:data" bottom title "equivalent drug-2 dose" left title "amount of disease" view
/* Now we use the Wilcoxon 2-sample test for paired data to compare our data
v with the corresponding predicted non-interaction response values r. */
wil2t(v,r)
[Wilcoxon 2 sample signed-rank test: are the medians of the
ranks of the observed paired data d1[] and d2[] plausibly equal?]
null hypothesis H0: median(d1) = median(d2).
The sum of positive ranks W-sample: T+ = 459.000000
The absolute sum of negative ranks W-sample: T- = 1819.000000
The probability P(W > 459.000000) = 0.999989
This means that a value of T+ at least as large as 459.000000 arises
about 99.998920 percent of the time, given H0.
The probability P(W > 1819.000000) = 0.000011
This means that a value of T- at least as large as 1819.000000 arises
about 0.001050 percent of the time, given H0.
The probability P[W > 1819.000000 or W < 459.000000] = 0.000022
This means that a rank-sum value at least as extreme as 1819.000000
arises about 0.002160 percent of the time, given H0.
: a 6 by 1 matrix TP
1: 67
2: 459
3: 1819
4: .999989202
5: 1.05008711E-5
6: 2.15957906E-5
In the wil2t output vector TP above, if the probability reported in TP[5] is small then we can reject the null hypothesis of equal responses, with probability TP[5] of being correct, and we can conclude that the alternate hypothesis: